Tutorial: Scanpy + Dask-AnnData¶
This notebook demonstrates converting a TileDB-SOMA ExperimentAxisQuery to an anndata whose X matrix is a Dask Array, and then running Scanpy’s distributed HVG and PCA
implementations on it.
In addition to dask, distributed, and tiledbsoma, this notebook requires dask-ml to be installed; here are some known-good pins:
pip install dask==2025.2.0 distributed==2025.2.0 dask-ml==2024.4.4 scanpy==1.11.0 scikit-learn==1.5.2
The first cell below contains Papermill parameters, which can be overridden when running this notebook programmatically, e.g.:
name=tutorial_scanpy_pca_dask
papermill -p tissue lung -p dask_chunk_size 200_000 $name.ipynb $name-lung_200k.ipynb
[1]:
from os import cpu_count
from psutil import virtual_memory
available_mem = virtual_memory().available
# Census
measurement_name = "RNA" # CELLxGENE Census measurement name
layer = "raw" # Read "raw" transcript counts
census = "2025-01-30" # Recent CELLxGENE Census version
tissue = "liver" # Query cells matching this "tissue_general" value
var_filter = None # Select all genes, by default
# Dask
dask_chunk_size = 100_000 # Number of "X" matrix rows per Dask task
worker_mem_allowance = 10 * 2**30 # 100k cell-rows per chunk uses about 10GiB in each worker
n_dask_procs = max(2, min( # Number of Dask worker processes to spawn
cpu_count(),
available_mem // worker_mem_allowance,
))
dask_disable_worker_mem_mgmt = True # Disable spilling and worker pausing/terminating; they tend to harm more than help
dask_graph_warn_size = "20MB" # Relax default (10MB) "Sending large graph" warning threshold ("distributed.admin.large-graph-warning-threshold" config key)
dashboard_port = 8787 # Port for the Dask web UI (for following jobs in real-time)
# Scanpy
hvg_inplace = True # Scanpy HVG: "inplace=True" by default
hvg_subset = False # Scanpy HVG: "subset=False" by default
Open experiment, initialize query¶
Open the CELLxGENE Census Experiment:
[2]:
import scanpy as sc
from somacore import AxisQuery
from tiledbsoma import Experiment, SOMATileDBContext
tiledb_config = {
"vfs.s3.no_sign_request": "true",
"vfs.s3.region": "us-west-2",
}
exp_uri = f"s3://cellxgene-census-public-us-west-2/cell-census/{census}/soma/census_data/homo_sapiens"
exp = Experiment.open(
exp_uri,
context=SOMATileDBContext(tiledb_config=tiledb_config),
)
exp
[2]:
<Experiment 's3://cellxgene-census-public-us-west-2/cell-census/2025-01-30/soma/census_data/homo_sapiens' (open for 'r') (2 items)
'ms': 's3://cellxgene-census-public-us-west-2/cell-census/2025-01-30/soma/census_data/homo_sapiens/ms' (unopened)
'obs': 's3://cellxgene-census-public-us-west-2/cell-census/2025-01-30/soma/census_data/homo_sapiens/obs' (unopened)>
Set up query for cells from the given tissue:
[3]:
%%time
obs_filter = "is_primary_data == True" # Deduplicate / only fetch primary representations of cells
if tissue:
obs_filter = f'tissue_general == "{tissue}" and {obs_filter}'
query = exp.axis_query(
measurement_name=measurement_name,
obs_query=AxisQuery(value_filter=obs_filter) if obs_filter else None,
var_query=AxisQuery(value_filter=var_filter) if var_filter else None,
)
CPU times: user 11.8 ms, sys: 3.04 ms, total: 14.8 ms
Wall time: 158 ms
Check the number of cells responsive to the query:
[4]:
%%time
query.n_obs
CPU times: user 4.05 s, sys: 1.6 s, total: 5.65 s
Wall time: 968 ms
[4]:
1429379
Initialize Dask¶
Create a cluster of local processes:
[5]:
import dask
import dask_ml # Used by `sc.pp.pca` later, fail early here if not installed # noqa: F401
from dask.distributed import Client, LocalCluster
if dask_disable_worker_mem_mgmt:
# Dask spilling and worker pause/terminate logic mostly interferes with our usage.
# It doesn't take into account available swap space and, once workers begin to spill
# or pause, the HVG/PCA jobs below never recover.
dask.config.set(
{
# "distributed.worker.memory.target": 0.7,
"distributed.worker.memory.spill": False,
"distributed.worker.memory.pause": False,
"distributed.worker.memory.terminate": False,
}
)
if dask_graph_warn_size:
# Relax default (10MB) "Sending large graph" warning threshold
dask.config.set({ "distributed.admin.large-graph-warning-threshold": dask_graph_warn_size })
if dask_chunk_size:
cluster = LocalCluster(
n_workers=n_dask_procs,
threads_per_worker=1, # Performance is better with just one thread per worker process
dashboard_address=f":{dashboard_port}",
)
print(f"{n_dask_procs=}")
client = Client(cluster)
else:
dask_chunk_size = None
client = None
client
n_dask_procs=24
[5]:
Client
Client-998e4379-1ffa-11f0-9ce2-0a61715a5721
| Connection method: Cluster object | Cluster type: distributed.LocalCluster |
| Dashboard: http://127.0.0.1:8787/status |
Cluster Info
LocalCluster
0dfede98
| Dashboard: http://127.0.0.1:8787/status | Workers: 24 |
| Total threads: 24 | Total memory: 246.55 GiB |
| Status: running | Using processes: True |
Scheduler Info
Scheduler
Scheduler-577fd058-b15b-4e3d-b2ce-4143fa04199c
| Comm: tcp://127.0.0.1:43111 | Workers: 24 |
| Dashboard: http://127.0.0.1:8787/status | Total threads: 24 |
| Started: Just now | Total memory: 246.55 GiB |
Workers
Worker: 0
| Comm: tcp://127.0.0.1:40945 | Total threads: 1 |
| Dashboard: http://127.0.0.1:34127/status | Memory: 10.27 GiB |
| Nanny: tcp://127.0.0.1:46219 | |
| Local directory: /tmp/dask-scratch-space/worker-z94t0m79 | |
Worker: 1
| Comm: tcp://127.0.0.1:45191 | Total threads: 1 |
| Dashboard: http://127.0.0.1:36411/status | Memory: 10.27 GiB |
| Nanny: tcp://127.0.0.1:32769 | |
| Local directory: /tmp/dask-scratch-space/worker-usp_kk6u | |
Worker: 2
| Comm: tcp://127.0.0.1:37477 | Total threads: 1 |
| Dashboard: http://127.0.0.1:41379/status | Memory: 10.27 GiB |
| Nanny: tcp://127.0.0.1:45997 | |
| Local directory: /tmp/dask-scratch-space/worker-dvsj6_q5 | |
Worker: 3
| Comm: tcp://127.0.0.1:42481 | Total threads: 1 |
| Dashboard: http://127.0.0.1:37229/status | Memory: 10.27 GiB |
| Nanny: tcp://127.0.0.1:44783 | |
| Local directory: /tmp/dask-scratch-space/worker-wuzxvsas | |
Worker: 4
| Comm: tcp://127.0.0.1:41441 | Total threads: 1 |
| Dashboard: http://127.0.0.1:35147/status | Memory: 10.27 GiB |
| Nanny: tcp://127.0.0.1:46479 | |
| Local directory: /tmp/dask-scratch-space/worker-wdr816w4 | |
Worker: 5
| Comm: tcp://127.0.0.1:38455 | Total threads: 1 |
| Dashboard: http://127.0.0.1:38631/status | Memory: 10.27 GiB |
| Nanny: tcp://127.0.0.1:37545 | |
| Local directory: /tmp/dask-scratch-space/worker-t5jjvncq | |
Worker: 6
| Comm: tcp://127.0.0.1:46499 | Total threads: 1 |
| Dashboard: http://127.0.0.1:41341/status | Memory: 10.27 GiB |
| Nanny: tcp://127.0.0.1:38935 | |
| Local directory: /tmp/dask-scratch-space/worker-rzh52kbq | |
Worker: 7
| Comm: tcp://127.0.0.1:46525 | Total threads: 1 |
| Dashboard: http://127.0.0.1:42759/status | Memory: 10.27 GiB |
| Nanny: tcp://127.0.0.1:33413 | |
| Local directory: /tmp/dask-scratch-space/worker-__vvbbr3 | |
Worker: 8
| Comm: tcp://127.0.0.1:35765 | Total threads: 1 |
| Dashboard: http://127.0.0.1:45609/status | Memory: 10.27 GiB |
| Nanny: tcp://127.0.0.1:36095 | |
| Local directory: /tmp/dask-scratch-space/worker-y_r996rt | |
Worker: 9
| Comm: tcp://127.0.0.1:41431 | Total threads: 1 |
| Dashboard: http://127.0.0.1:33101/status | Memory: 10.27 GiB |
| Nanny: tcp://127.0.0.1:37301 | |
| Local directory: /tmp/dask-scratch-space/worker-r3lgcszf | |
Worker: 10
| Comm: tcp://127.0.0.1:41223 | Total threads: 1 |
| Dashboard: http://127.0.0.1:38683/status | Memory: 10.27 GiB |
| Nanny: tcp://127.0.0.1:46385 | |
| Local directory: /tmp/dask-scratch-space/worker-dqsy8hpz | |
Worker: 11
| Comm: tcp://127.0.0.1:32917 | Total threads: 1 |
| Dashboard: http://127.0.0.1:39351/status | Memory: 10.27 GiB |
| Nanny: tcp://127.0.0.1:44559 | |
| Local directory: /tmp/dask-scratch-space/worker-9269pmbe | |
Worker: 12
| Comm: tcp://127.0.0.1:46691 | Total threads: 1 |
| Dashboard: http://127.0.0.1:39097/status | Memory: 10.27 GiB |
| Nanny: tcp://127.0.0.1:46613 | |
| Local directory: /tmp/dask-scratch-space/worker-bemmhqfq | |
Worker: 13
| Comm: tcp://127.0.0.1:39711 | Total threads: 1 |
| Dashboard: http://127.0.0.1:38153/status | Memory: 10.27 GiB |
| Nanny: tcp://127.0.0.1:41581 | |
| Local directory: /tmp/dask-scratch-space/worker-byvs2oin | |
Worker: 14
| Comm: tcp://127.0.0.1:41949 | Total threads: 1 |
| Dashboard: http://127.0.0.1:36143/status | Memory: 10.27 GiB |
| Nanny: tcp://127.0.0.1:38979 | |
| Local directory: /tmp/dask-scratch-space/worker-9_8z3l8l | |
Worker: 15
| Comm: tcp://127.0.0.1:46179 | Total threads: 1 |
| Dashboard: http://127.0.0.1:44059/status | Memory: 10.27 GiB |
| Nanny: tcp://127.0.0.1:35137 | |
| Local directory: /tmp/dask-scratch-space/worker-rg5uies3 | |
Worker: 16
| Comm: tcp://127.0.0.1:33403 | Total threads: 1 |
| Dashboard: http://127.0.0.1:45875/status | Memory: 10.27 GiB |
| Nanny: tcp://127.0.0.1:36437 | |
| Local directory: /tmp/dask-scratch-space/worker-z89clwwc | |
Worker: 17
| Comm: tcp://127.0.0.1:35123 | Total threads: 1 |
| Dashboard: http://127.0.0.1:40691/status | Memory: 10.27 GiB |
| Nanny: tcp://127.0.0.1:44181 | |
| Local directory: /tmp/dask-scratch-space/worker-7bibqf_g | |
Worker: 18
| Comm: tcp://127.0.0.1:43439 | Total threads: 1 |
| Dashboard: http://127.0.0.1:37283/status | Memory: 10.27 GiB |
| Nanny: tcp://127.0.0.1:37295 | |
| Local directory: /tmp/dask-scratch-space/worker-8kmfmv3a | |
Worker: 19
| Comm: tcp://127.0.0.1:39637 | Total threads: 1 |
| Dashboard: http://127.0.0.1:39425/status | Memory: 10.27 GiB |
| Nanny: tcp://127.0.0.1:45493 | |
| Local directory: /tmp/dask-scratch-space/worker-8n7la9o9 | |
Worker: 20
| Comm: tcp://127.0.0.1:38459 | Total threads: 1 |
| Dashboard: http://127.0.0.1:39473/status | Memory: 10.27 GiB |
| Nanny: tcp://127.0.0.1:33257 | |
| Local directory: /tmp/dask-scratch-space/worker-3dsw_5l2 | |
Worker: 21
| Comm: tcp://127.0.0.1:36157 | Total threads: 1 |
| Dashboard: http://127.0.0.1:38073/status | Memory: 10.27 GiB |
| Nanny: tcp://127.0.0.1:39563 | |
| Local directory: /tmp/dask-scratch-space/worker-0uibf76k | |
Worker: 22
| Comm: tcp://127.0.0.1:38407 | Total threads: 1 |
| Dashboard: http://127.0.0.1:35439/status | Memory: 10.27 GiB |
| Nanny: tcp://127.0.0.1:45877 | |
| Local directory: /tmp/dask-scratch-space/worker-3inbdix2 | |
Worker: 23
| Comm: tcp://127.0.0.1:36347 | Total threads: 1 |
| Dashboard: http://127.0.0.1:36625/status | Memory: 10.27 GiB |
| Nanny: tcp://127.0.0.1:42413 | |
| Local directory: /tmp/dask-scratch-space/worker-xs6pqvir | |
It’s recommended to use one worker thread per Dask process (threads_per_worker=1 above). Performance (wall-clock and memory use) suffers when more than one Dask worker thread runs in the same Dask worker process.
It’s also encouraged to open the dashboard at 127.0.0.1:8787/status, to follow along with tasks, progress, and memory use in real-time. Here’s an example screenshot of that UI, from a completed run of this notebook:
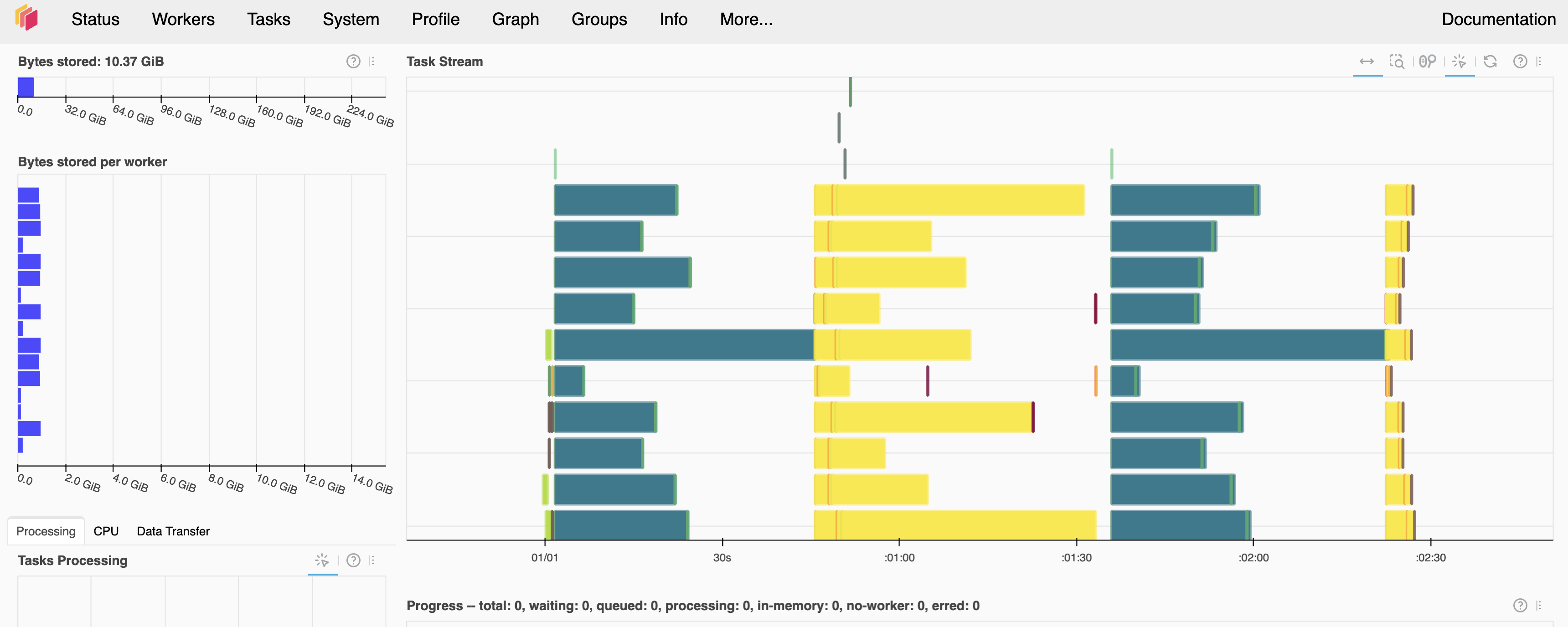
Convert query to Dask-backed anndata¶
[6]:
%%time
adata = query.to_anndata(
layer,
dask=dict(
chunk_size=dask_chunk_size,
tiledb_config=tiledb_config,
),
)
adata
CPU times: user 10.5 s, sys: 9.76 s, total: 20.2 s
Wall time: 10.1 s
[6]:
AnnData object with n_obs × n_vars = 1429379 × 61888
obs: 'soma_joinid', 'dataset_id', 'assay', 'assay_ontology_term_id', 'cell_type', 'cell_type_ontology_term_id', 'development_stage', 'development_stage_ontology_term_id', 'disease', 'disease_ontology_term_id', 'donor_id', 'is_primary_data', 'observation_joinid', 'self_reported_ethnicity', 'self_reported_ethnicity_ontology_term_id', 'sex', 'sex_ontology_term_id', 'suspension_type', 'tissue', 'tissue_ontology_term_id', 'tissue_type', 'tissue_general', 'tissue_general_ontology_term_id', 'raw_sum', 'nnz', 'raw_mean_nnz', 'raw_variance_nnz', 'n_measured_vars'
var: 'soma_joinid', 'feature_id', 'feature_name', 'feature_type', 'feature_length', 'nnz', 'n_measured_obs'
The X matrix is a Dask Array:
[7]:
print(type(adata.X))
adata.X
<class 'dask.array.core.Array'>
[7]:
|
|||||||||||||
Scanpy preprocessing¶
PCA uses normalized cell counts, and log1p values:
[8]:
%%time
sc.pp.normalize_total(adata)
CPU times: user 23.4 ms, sys: 8.21 ms, total: 31.6 ms
Wall time: 25 ms
[9]:
%%time
sc.pp.log1p(adata)
CPU times: user 4.45 ms, sys: 3.22 ms, total: 7.67 ms
Wall time: 9.49 ms
Scanpy HVG¶
[10]:
%%time
hvg = sc.pp.highly_variable_genes(adata, inplace=hvg_inplace, subset=hvg_subset)
hvg
CPU times: user 7.97 s, sys: 2.03 s, total: 10 s
Wall time: 32.1 s
Scanpy PCA¶
NOTE: it’s important to run HVG before PCA, so that PCA will only operate on the highly-variable genes. Otherwise, it will run on all ≈60k genes, and use intractable amounts of memory and compute.
[11]:
%%time
sc.pp.pca(adata)
CPU times: user 1min 8s, sys: 3.99 s, total: 1min 12s
Wall time: 1min 4s
[12]:
%%time
adata.obsm["X_pca"] = adata.obsm["X_pca"].compute()
CPU times: user 11.7 s, sys: 2.6 s, total: 14.3 s
Wall time: 23.5 s
Subset cell types, plot¶
[13]:
t10 = adata.obs.cell_type.value_counts().iloc[:10]
a10 = adata[adata.obs.cell_type.isin(t10.index)]
a10
[13]:
View of AnnData object with n_obs × n_vars = 893703 × 61888
obs: 'soma_joinid', 'dataset_id', 'assay', 'assay_ontology_term_id', 'cell_type', 'cell_type_ontology_term_id', 'development_stage', 'development_stage_ontology_term_id', 'disease', 'disease_ontology_term_id', 'donor_id', 'is_primary_data', 'observation_joinid', 'self_reported_ethnicity', 'self_reported_ethnicity_ontology_term_id', 'sex', 'sex_ontology_term_id', 'suspension_type', 'tissue', 'tissue_ontology_term_id', 'tissue_type', 'tissue_general', 'tissue_general_ontology_term_id', 'raw_sum', 'nnz', 'raw_mean_nnz', 'raw_variance_nnz', 'n_measured_vars'
var: 'soma_joinid', 'feature_id', 'feature_name', 'feature_type', 'feature_length', 'nnz', 'n_measured_obs', 'highly_variable', 'means', 'dispersions', 'dispersions_norm'
uns: 'log1p', 'hvg', 'pca'
obsm: 'X_pca'
varm: 'PCs'
[14]:
%%time
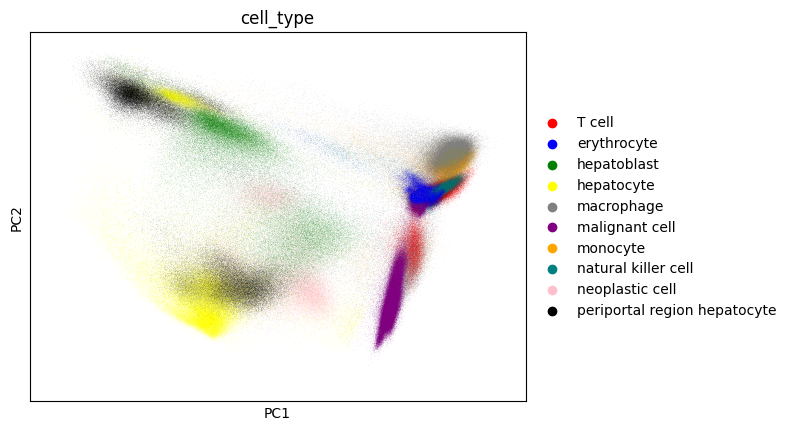
sc.pl.pca(a10, color="cell_type", palette=["red", "blue", "green", "yellow", "grey", "purple", "orange", "teal", "pink", "black"])
/home/ubuntu/.pyenv/versions/3.11.8/lib/python3.11/site-packages/anndata/_core/anndata.py:1146: ImplicitModificationWarning: Trying to modify attribute `.var` of view, initializing view as actual.
df[key] = c
CPU times: user 5.74 s, sys: 300 ms, total: 6.04 s
Wall time: 5.77 s